
Dr. Shruthi Viswanath - Research
Integrative structure determination
Structures of several large protein complexes and assemblies are difficult to obtain using traditional experimental methods. Integrative structure determination fills this gap; various types of experimental data are combined along with principles from physics, statistical inference, and prior models to obtain the structure. The different sources of input information may span multiple scales (for example, X-ray data is at the atomic scale, while FRET distances are at the domain scale). However, these various sources can provide complementary information (for example, EM maps may provide the shape of a complex while chemical crosslinks may provide the orientation of binding interfaces). We have used structural, biochemical, biophysical, cell biological, genetic, and in-silico bioinformatics information for deducing the structure of assemblies. Of late, AI-based methods have enabled amazing advances in structural biology and it is an exciting field. For us, simulation and analysis of large macromolecular assemblies leads us to interesting opportunities for developing new modeling methods. For more information, please read two recent reviews on integrative structural modeling from our group.
We recently determined the integrative structures of the Nucleosome Remodeling and Deacetylase (NuRD) complex, a chromatin-modifying assembly that regulates gene expression and DNA damage repair (PDB 9A8C), and the desmosomal outer dense plaque, an assembly that mediates cell-cell adhesion and signaling (PDB 9A8U).
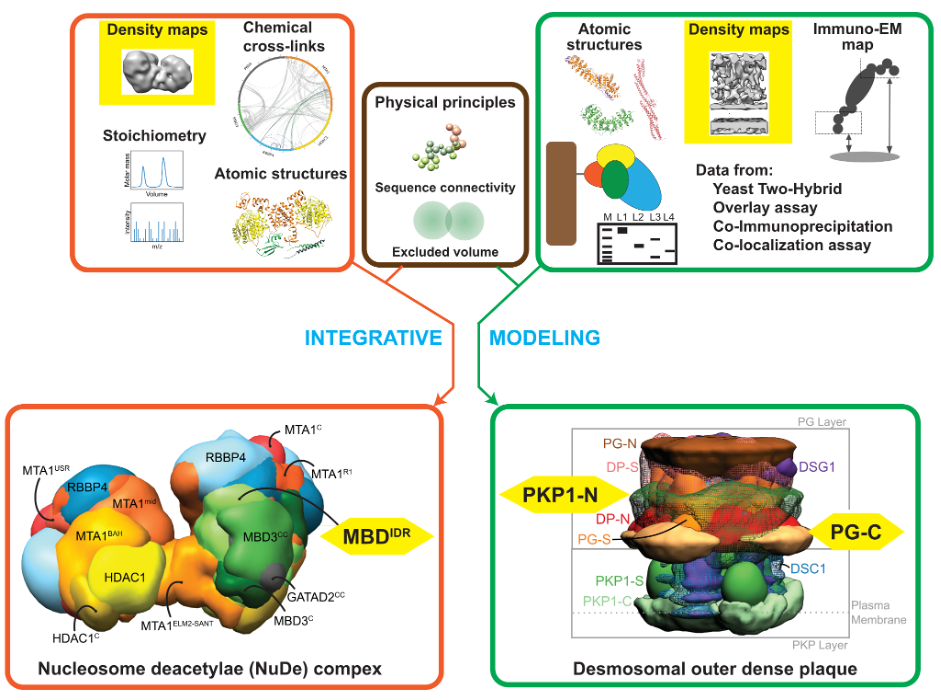
Schematic describing integrative structure determination for the nucleosome remodeling and deacetylase complex (orange box) and the desmosomal outer dense plaque (green box) combining data from multiple sources. Low-resolution cryo-EM and cryo-ET maps (yellow) and intrinsically disordered regions (yellow) in both complexes are highlighted as emerging areas for method development.
Two recurrent modeling challenges were noticed across a range of studies such as the two mentioned above. One was the need to develop methods for incorporating disordered regions in these assemblies and another was to better utilize information from cryo-electron tomography, a timely challenge as structural biology is moving towards in situ characterization. We are currently focusing our method development in these two areas.
In our lab, we have also developed several methods across the spectrum of integrative modeling. For example, our method to annotate precision for integrative structural models is now part of the PDB validation pipeline.
Please look at our lab website for further details.