
Prof. Jayant B. Udgaonkar - Research
How do proteins fold, unfold and misfold?
The polypeptide chain of a protein must bend, loop, coil, turn and twist itself in a very precise manner while folding into the unique structure that enables the protein to function in the cell. The protein folding problem is to understand how structure develops as a protein folds. It has been a long-standing, unsolved puzzle in biology, whose solution has obvious biotechnological as well as medical implications. In particular, the improper folding of some proteins, and their consequent aggregation into amyloid fibrils, are characteristic features of several neuro-degenerative diseases as well as of the prion diseases. An understanding of the mechanism of protein folding will also lead to a better understanding of the other facet of the protein folding problem, which is how to predict the functional structure of a protein from the amino-acid sequence that specifies it.
My laboratory uses several small proteins, including monellin, the SH3 domain of the PI3- kinase, barstar, tau, and the mouse prion protein as archetypical model proteins for studying how proteins fold, unfold as well as aggregate. We also study how correct folding is assisted by the chaperone GroEL. We use the tools of protein engineering and physical biochemistry. These include diverse optical spectroscopic methods such as time-resolved fluorescence, as well as mass spectrometry and nuclear magnetic resonance spectroscopy. Our kinetic measurements span the time domain of 40 microseconds to 10 hours.
Highlights of our recent work on protein folding and unfolding include (1) the demonstration that multiple pathways are available for the folding and unfolding of monellin and that switching between alternative pathways can occur with a change in folding or unfolding conditions; (2) the demonstration that the native state of the prion protein undergoes unusually high fluctuations, which makes it extremely malleable to structural change; (3) the demonstration that protein unfolding reactions can occur through intermediates with non-native interactions; and (4) the demonstration that protein unfolding proceeds through dry and wet globules and a solvated transition state. Highlights of our recent work on protein misfolding and aggregation include (1) the structural characterization of oligomeric intermediates and worm-like fibrils formed by the mouse prion protein, and the demonstration that these aggregates can perturb membrane structure; (2) the temporal dissection of the structural changes that occur during fibril formation by the prion protein; and (3) the demonstration of a secondary pathway for fibril formation by the tau protein, and the delineation of structural changes that occur during fibril formation.

Selected publications:
- Singh, J., Sabareesana, A.T, Mathew, M.K. and Udgaonkar, J.B. (2012). Development of the structural core and of conformational heterogeneity during the conversion of oligomers of the mouse prion protein toworm-like amyloid fibrils. J. Mol. Biol. 423, 217-231.
- Udgaonkar, J.B. (2013). Polypeptide chain collapse and protein folding. Arch. Biochem. Biophys. 531, 24-33.
- Ramachandran, G. and Udgaonkar, J.B. (2013) Mechanistic studies unravel the complexity inherentin tau aggregation leading to Alzheimer’s disease and the tauopathies. Biochemistry 52, 4107-4126.
1 THE UTILIZATION OF ALTERNATIVE PATHWAYS DURING PROTEIN FOLDING AND UNFOLDING
Nilesh Aghera
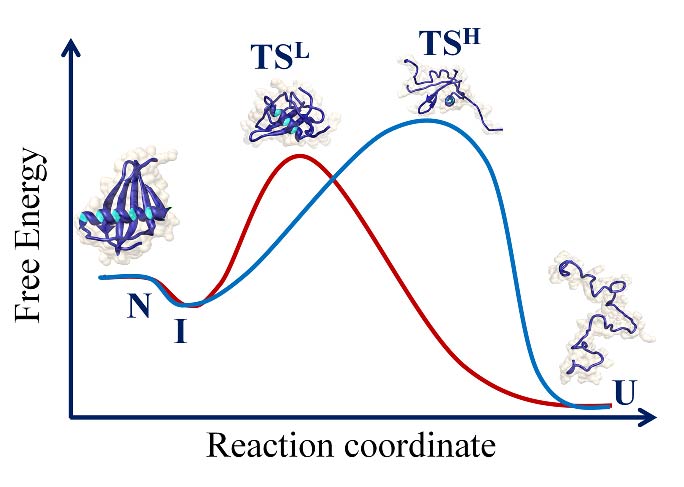
Determining whether a protein utilizes multiple pathways to fold and unfold is an important goal in protein folding studies. Multiple pathways may manifest themselves by causing the dependence on denaturant concentration of the logarithm of the observed rate constant of folding and/or unfolding to have an upward curvature. This will happen when the transition states on the alternative pathways differ in their compactness and structure but not significantly in energy. Detection of multiple pathways in this manner is rare.
Upward curvatures were detected in the chevron arms for both the fast and slow phases of refolding of heterodimeric monellin. It was shown that the initial encounter complex formed by the two chains folds via two folding routes, one of which is populated by a productive folding intermediate. Similarly an upward curvature was observed for the unfolding of single chain monellin. For both proteins, it was shown that the folding or unfolding reaction switches from one pathway to another with a change in denaturant concentration.
2 COMPLEXITY OF THE FOLDING PATHWAY OF THE SH3 DOMAIN OF PI3 KINASE
Amrita Dasgupta
Protein folding reactions often appear to be deceptively simple. The hidden complexity of the folding reaction of the apparently “two-state” folder, the SH3 domain of PI3 kinase (PI3K- SH3 domain) was revealed by the addition of a co-solute that stabilized folding intermediates. A collapsed intermediate before the rate limiting step and a more unfolded like intermediate after the rate limiting step were identified in kinetic folding and unfolding studies. A four-state mechanism for folding and unfolding was shown to be valid over the entire range of denaturant concentration studied. Very interestingly, the intermediate that forms after the rate limiting step of folding was shown to possess non-native structure.
3 CRITICAL EVALUATION OF A TWO-STATE MODEL FOR EQUILIBRIUM UNFOLDING
Megha Kishore
The probes commonly used to measure equilibrium unfolding reactions of proteins cannot detect the underlying heterogeneity inherent in the reactions, and hence these reactions are described as two-state. Time resolved fluorescence resonance energy transfer (TRFRET) measurements revealed the gradual expansion of the native state of the PI3K-SH3 domain at low denaturant concentration, and that the unfolding cannot be described by models that invoke only a few discrete states.
4 THE NATIVE STATE OF THE PRION PROTEIN HAS A CONFORMATIONALLY FLEXIBLE AND MALLEABLE NATIVE STATE
Roumita Moulick
The prion protein appears to be unusually susceptible to conformational change. Unlike most other proteins it can easily adopt alternative misfolded conformations. A complete thermodynamic characterization of the unfolding of the mouse prion protein revealed, by measurement of heat capacity changes, that the native state undergoes substantial fluctuations in enthalpy and hence, in structure.
5 HIGH ENERGY INTERMEDIATES IN PROTEIN UNFOLDING
Pooja Malhotra
A protein unfolding reaction usually appears to be so dominated by a large free energy barrier, that identifying and characterizing high energy intermediates, and hence dissecting the unfolding reaction into multiple structural transitions, has proven to be a challenge. Native-state thiol labeling measurements were used to identify high energy intermediates as well as to delineate the barriers to the disruption of side-chain packing interactions and to site-specific solvent exposure in different regions of the small protein monellin.
6 INITIAL CHAIN COLLAPSE DURING THE FOLDING OF MONELLIN
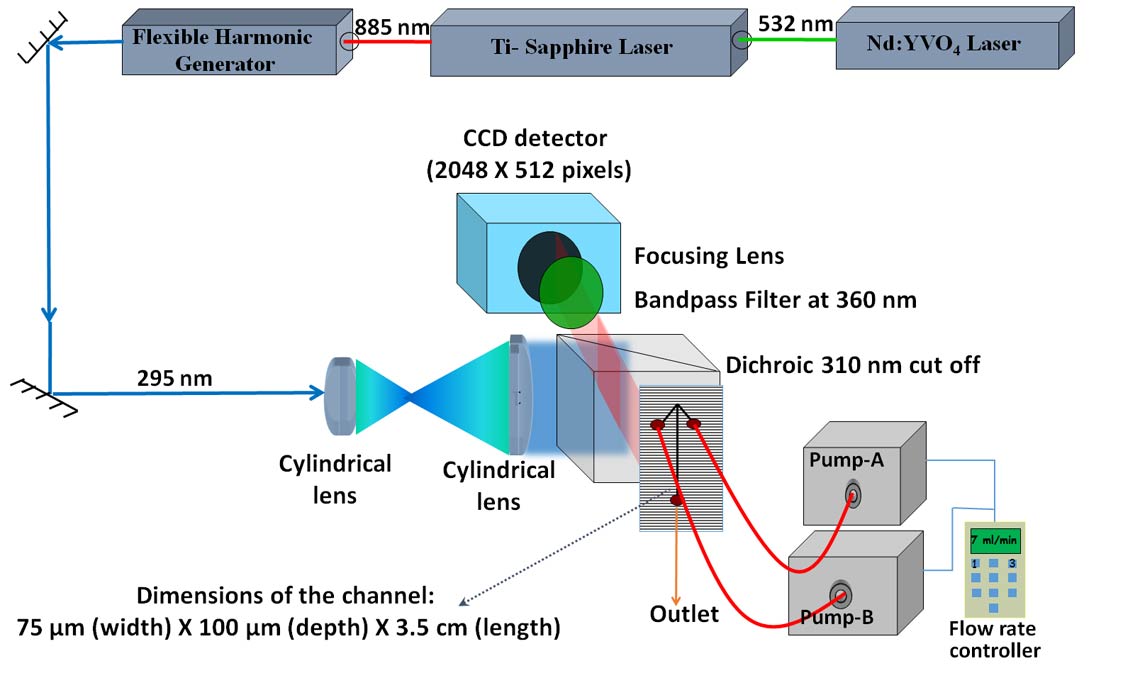
G. Rama Reddy
A microsecond mixer capable of measuring protein folding rates as high as 25,000 s-1 was developed. Initial use of the mixer has shown that different regions of the protein chain undergo initial chain collapse at different rates independent of each other.
7 INITIAL STRUCTURAL CHANGES DURING THE UNFOLDING OF THE PI3K-SH3 DOMAIN
Prashant Jethva
Hydrogen exchange coupled to mass spectrometry (HX-MS) is being used to compare the unfolding of the PI3K-SH3 domain in two denaturants. Initial results showed that in the absence of denaturant, the native state transforms into an intermediate in a gradual and not in an all-or- none manner.
8 GROEL-ASSISTED REFOLDING OF HETERODIMERIC MONELLIN
Neha Nandwani
A study of the mechanism by which the chaperone GroEL assists the folding of the heterodimeric protein monellin, was begun.
9 STRUCTURAL CHARACTERIZATION OF THE TRANSFORMATION OF OLIGOMERS OF THE MOUSE PRION PROTEIN INTO WORM-LIKE AMYLOID FIBRILS
Jogender Singh and A.T. Sabareesan, in collaboration with M.K. Mathew
Understanding how structure develops and conformational heterogeneity manifests itself during the course of amyloid fibril formation by the prion protein is important for understanding prion diseases. HX-MS was used to delineate the structural cores of two oligomers formed by the mouse prion protein at low pH, and to understand in structural terms why only the larger oligomer and not the smaller can transform into worm-like fibrils. All three amyloid aggregates were shown to be capable of disrupting lipid membrane structure, pointing to a mechanism by which they may be toxic.
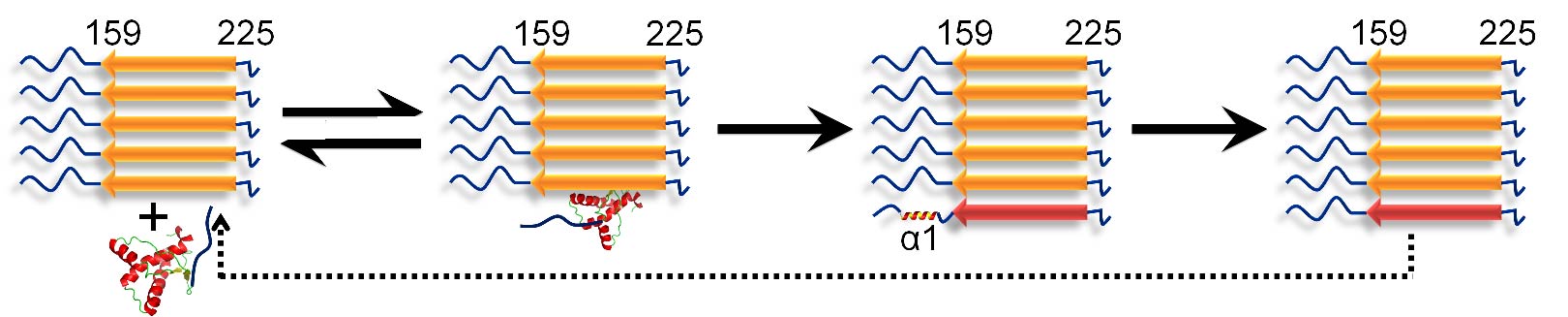
10 STRUCTURAL CHARACTERIZATION OF THE CONVERSION OF MONOMERIC PRION PROTEIN INTO AMYLOID FIBRILS
Jogender Singh
A molecular understanding of prion diseases requires an understanding of the mechanism of amyloid fibril formation by the prion protein. HX-MS was used to describe the conformational conversion of monomeric mouse prion protein into aggregated amyloid fibrils. It was shown that conformational conversion occurs in two steps after the binding of monomer to fibril, with helix 1 unfolding only after helices 2 and 3 transform into beta-sheet.
11 MECHANISM OF AMYLOID FIBRIL FORMATION BY HUMAN TAU AND STRUCTURAL CHARACTERIZATION OF TAU FIBRILS
Gayathri Ramachandran
Tau protein is an intrinsically disordered protein which is known to aggregate and form neurofibrillary tangles in Alzheimer’s disease as well as in a wide range of other taupathies. The polyanion heparin is commonly used as an inducer in studies of tau aggregation in vitro, but its kinetic role in inducing aggregation had not been known. Similarly the kinetic role of intermediates populated during tau aggregation was not understood. Amyloid fibril formation by tau4RD, a tau four-repeat domain construct, in the presence of the inducer heparin was shown to occur by a nucleation dependent polymerization mechanism, and the kinetic role of heparin was shown to be restricted to nucleation. It was shown that rod-like protofibrils were populated during the course of aggregation but that these were off the pathway of fibril formation. The amyloid fibril formation reaction of tauK18, another tau four-repeat construct was also studied. It was shown that not only is there a primary nucleation pathway but also a secondary pathway for fibril growth. It was shownthat the dominant secondary pathway is fibril fragmentation.
HX-MS was used to show that the fibrils formed by tau4RD and tauK18 are constructed on similar structural principles, but that the tauK18 fibril has a slightly more stable core.
12 CELLULAR MODEL OF POLYGLUTAMINE AGGREGATION
Vishal Bhardwaj, in collaboration with M.M. Panicker
The aggregation of poly-glutamine rich proteins in inclusion bodies in cells is closely linked with numerous neuro-degenerative disorders, but the organization of protein in the inclusions is poorly understood. Fluorescence anisotropy based measurements were used to probe packing in inclusions at various stages of growth in live cells. It was shown, by measurement of the enhancement of the homo-FRET that accompanies inclusion growth, that the protein packing evolves and becomes tighter as the inclusion grows in size.
13 AMYLOID FIBRIL FORMATION BY THE CHAIN B SUBUNIT OF MONELLIN
A.T. Sabareesan
Proteins possessing very different structures, or even no structure, form fibrils that are very similar in internal structure, suggesting that they aggregate by similar mechanisms. The aggregation reaction of chain B of monellin was shown to meet all the stringent kinetic criteria of a nucleation dependent polymerization mechanism valid over a wide range of protein concentration. Off- pathway spherical oligomers were shown to form transiently during fibril formation.
14 ARCHITECTURE OF PRION FIBRILS
Ishita Sengupta
A study of the morphology of prion fibrils, using TRFRET spectroscopy as well as NMR spectroscopy was initiated.
15 AMYLOID FIBRIL FORMATION BY α-SYNUCLEIN
Pratibha Kumari
A study of the mechanism by which the dye thioflavin T modulates amyloid fibril formation by α-synuclein was begun.