
Prof. Sumantra Chattarji
I. Summary
Memories come in many different flavors, some more potent than others. Emotionally significant experiences tend to be well remembered, and the amygdala has a pivotal role in this process. But the rapid and efficient encoding of emotional memories can become maladaptive — severe stress often turns them into a source of chronic anxiety. What are the cellular and circuit mechanisms underlying these powerful emotional symptoms? To answer this question, we have been using a range of behavioral, morphometric, in vitro and in vivo electrophysiological, and molecular tools to identify neural correlates of stress-induced modulation of amygdalar structure and function — from cellular and synaptic mechanisms to their behavioral consequences in rodents. Our findings point to unique features of stress-induced plasticity in the amygdala, which are in striking contrast to those seen in the hippocampus and prefrontal cortex, and could have long-term consequences for pathological fear and anxiety observed in individuals suffering from affective disorders (Chattarji et al., Nature Neuroscience, 2015).
In addition to behavioral experience, the genes we inherit can also cause cognitive and emotional dysfunction. Notably, individuals afflicted with certain types of autism spectrum disorder often exhibit impaired cognitive function alongside high anxiety and mood lability. Hence, we are extending our analyses to human neural stem cells, as well new rat models, to identify circuit, cellular and molecular targets that can be used to correct symptoms of Fragile X Syndrome, the leading genetic cause of autism.
II. Current research and new directions
1. Effects of stress distributed across neural networks: interactions between brain areas
As discussed earlier, animal studies show that stress elicits divergent patterns of plasticity across brain regions. For instance, repeated stress causes loss of dendrites and spines in the medial prefrontal cortex (mPFC). In the basolateral amygdala (BLA), by contrast, chronic stress strengthens the structural basis of synaptic connectivity through dendritic growth and spinogenesis. Physiological and molecular measures of synaptic plasticity also exhibit these contrasting features. As useful as these studies have been in examining the effects of stress across biological scales, much of this evidence was gathered from postmortem analyses. Less is known about how stress affects neural activity in the intact brain of behaving animals. Further, in many of these studies, stress-induced plasticity was viewed as stand-alone effects intrinsic to individual brain areas, despite extensive interconnections between them. Indeed, interactions between these brain areas together give rise to behaviors that are affected by stress. One such behavior involves the expression of fear memories, various facets of which depend on both the BLA and mPFC.
Stress is known to exert its detrimental effects not only by enhancing fear, but also by impairing its extinction. However, in earlier studies stress exposure preceded both processes. Thus, compared to unstressed animals, stressed animals had to extinguish fear memories that were strengthened by prior exposure to stress. Here we dissociate the two processes to examine if stress specifically impairs the acquisition and recall of fear extinction. Strikingly, when fear memories were formed before stress exposure, thereby allowing animals to initiate extinction from comparable levels of fear, recall of fear extinction was unaffected. Despite this we observed a persistent increase in theta activity in the BLA. Theta activity in the mPFC, by contrast, was normal. Stress also disrupted mPFC-BLA theta-frequency synchrony and directional coupling. Finally, pharmacological inactivation of the BLA did not affect recall or extinction of fear in these animals. Thus, in the absence of the fear-enhancing effects of stress, the expression of fear during and after extinction reflects normal regulation of theta activity in the mPFC, not theta hyperactivity in the amygdala (Rahman et al., eLife, 2018).
2. Beyond neurons: contrasting effects of stress on astrocytes in the amygdala versus hippocampus
Stress causes divergent patterns of structural and physiological plasticity in the hippocampus versus amygdala (Chattarji et al., Nature Neurosci., 2015). However, a majority of earlier studies focused primarily on neurons. Despite growing evidence for the importance of glia in health and disease, relatively little is known about how stress affects astrocytes. Hence, we are examining the impact of chronic immobilization stress (2h/day, 10 days), on the number and structure of astrocytes in the rat hippocampus and amygdala. We observe a reduction in the number of GFAP-positive astrocytes in the basal amygdala (BA) a day after the end of chronic stress. Detailed morphometric analysis of individual dye-filled astrocytes also reveals a decrease in the neuropil volume occupied by these astrocytes in the BA. By contrast, the same chronic stress has no effect on the number or morphology of astrocytes in hippocampal area CA3. Finally, we confirm previous reports that chronic stress triggered dendritic growth in BA principal neurons. Notably, BA neurons exhibiting dendritic hypertrophy are located adjacent to astrocytes that had undergone atrophy, thereby providing striking examples of the opposite effects of the same stress on neurons versus astrocytes. Together, these findings offer new evidence that the morphological effects of stress not only vary with brain regions, but can also cause markedly different effects on neurons versus astrocytes within the same brain area. This, in turn, sets the stage for exploring the potential role of astrocytes, and their glutamate reuptake machinery, in the contrasting effects of stress on the amygdala versus hippocampus.
3. Even a positive experience, like environmental enrichment, elicits contrasting effects in the amygdala and hippocampus
As described earlier, an aversive experience like chronic stress strengthens fear memories, causes dendritic growth and spinogenesis, and enhances LTP in the amygdala. Paradoxically, similar effects are seen in the hippocampus and cortex, not as a result of stress, but after a positive experience like environmental enrichment (EE) consisting of increased exercise, social interactions and exposure to objects that encourage exploration. This raises an intriguing question – how would the amygdala respond to EE? If EE elicits cellular changes in the amygdala similar to the hippocampus, then our earlier results predict EE to enhance anxiety and fear learning. But one expects positive experiences to reduce, not increase, fear and anxiety. On the other hand, if EE does reduce fear and anxiety, would it induce cellular changes in the amygdala that are different from the hippocampus? Indeed, we find that EE decreases amygdala-dependent auditory fear conditioning, as well as anxiety. But, EE does not affect spine density in the LA. In contrast, the same EE enhances hippocampal spine density and contextual fear conditioning (Malik & Chattarji, J. Neurophysiol., 2012). Consistent with these behavioral and morphological changes, whole-cell recordings in hippocampal slices show EE to enhance LTP and mEPSC frequency. In striking contrast, EE elicits a reduction in mEPSC frequency, along with smaller magnitude of LTP in the LA. Further, the EE-induced reduction in LTP in the LA is due to a decrease in pre-synaptic release of glutamate at thalamic inputs. Together these findings suggest that plasticity elicited by a positive experience also appears to be different in the amygdala. We are now examining if prior exposure to EE can prevent the detrimental effects of chronic stress in the amygdala and hippocampus.
In conclusion, research in my laboratory has shown that the amygdala has unique features of structural and functional plasticity that make it especially relevant to our understanding of the pathological consequences of severe and repeated stress. Furthermore, whatever happens in the amygdala seems to occur in concert with functional changes in other, interconnected brain regions. Therefore, a major goal of future studies in my laboratory will be to investigate the role of the amygdala in stress effects distributed across wider neural networks including the hippocampus and mPFC. This will not only help us to understand the complex interactions between stress and memory, but may also reveal common targets for therapeutic interventions against the diverse emotional and cognitive symptoms of stress-related psychiatric disorders.
III. A major international initiative in translational neuroscience
1. Centre for Brain Development and Repair (CBDR)
Finally, some of our exciting research on Fragile X Syndrome (FXS), the leading genetic cause of autism, have emerged from a line of investigation that has been a relatively small and recent in my lab. However, the novelty of our findings has led to a major international collaborative partnership with several well-known neuroscientists, including Sir Adrian Bird, Richard Morris, Siddharthan Chandran, Peter Kind, and others at the University of Edinburgh, UK. This has culminated in the formation of the Centre for Brain Development and Repair (CBDR), funded by a major grant from the Department of Biotechnology (DBT), that I founded in 2014 and now head. This international collaborative center, the first of its kind in India, combines a range of expertise in several fields of neurobiology, including new transgenic rat models and human induced pluripotent stem cell (iPSC) based in vitro systems, with the initial goal of investigating autism spectrum disorders and intellectual disabilities (ASD/IDs).
Over the past four years at CBDR, we have succeeded in generating and characterizing electrophysiological properties of cortical neurons and astrocytes derived from human iPSCs (for comparing multiple lines of normal and patient derived cells). Further, we have also established 9 new transgenic rat models (FXS, Syngap, PTEN, Rett Syndrome (MeCP2), Neurexin 1, Cntnap2, Cdkl-5 and NR2A) of highly penetrant single-gene causes of ASD/IDs to better model autistic and cognitive behaviors that can accurately reflect autistic features in humans. This is the single largest collection of new rat models of ASD/IDs in the world.
We are now carrying out detailed comparative assessments of these new models simultaneously at the behavioral, electrophysiological and biochemical levels across multiple brain areas (amygdala, hippocampus, medial prefrontal cortex) and developmental time points. Together these complementary strategies, spanning both animal and human neural stem cell models, seek to identify convergent signatures of ASD/ID, which once found can then be addressed to design rationale-based, reasonable therapies for these disorders.
The framework that has emerged from our work at CBDR is now providing new mechanistic insights into the debilitating emotional symptoms associated with ASD/IDs. For instance, group I metabotropic glutamate receptor subtype mGluR5 has been a major focus of research on the pathophysiology of FXS. Although mGluR5-antagonists prevent fear and anxiety, major symptoms of ASD/IDs, little is known about how the same receptor in the amygdala gives rise to both. Combining in vitro and in vivo activation of mGluR5 in rats, we identify (Rahman et al., eLife, 2017) specific changes in intrinsic excitability and synaptic plasticity in basolateral amygdala neurons that give rise to temporally distinct and mutually exclusive effects on fear-related behaviors. The immediate impact of mGluR5 activation is to produce anxiety manifested as indiscriminate fear of both tone and context. Surprisingly, this state does not interfere with the proper encoding of tone-shock associations that eventually lead to enhanced cue-specific fear. These results provide a new framework for dissecting the functional impact of amygdalar mGluR-plasticity on fear versus anxiety in health and disease.
IV. Key findings from earlier work
1. Stress, Memory and the Amygdala
1.1 Divergent patterns of stress-induced plasticity across brain regions
After establishing my new laboratory in 1999 at the National Centre for Biological Sciences (NCBS), I decided to focus on a key question about memory mechanisms in the brain that had fascinated me for quite some time. Unlike memories of facts and events encoded by the hippocampus, emotional memories can be established rapidly and are often more potent and persistent. Why are emotional memories more powerful than memories of facts and events? The mechanisms underlying this contrast were not well understood when I started my lab at NCBS. Therefore, the initial focus of my research was on investigating the neural basis of this contrast at multiple levels of organization.
An effective strategy to probe the differences between amygdalar and hippocampal plasticity comes from behavioral studies demonstrating the divergent effects of stress on the memory output of these two brain structures. In rodents, severe and repeated stress has been shown to cause hippocampal dendritic atrophy, impaired hippocampal synaptic plasticity and learning. Moreover, rodent models of stress-induced neuronal atrophy may provide one potential explanation of the loss of hippocampal volume and cognitive deficits associated with stress-related psychiatric disorders, such as post-traumatic stress disorder (PTSD), major depression, etc. In contrast, stress amplifies fear and anxiety related behavior that depends on the amygdala. However, when we started our work, not much was known about the cellular basis of the enhancing effects of stress on amygdalar function. Thus, stress provided an ideal “top-down” behavioral perturbation to investigate factors accounting for the striking differences between hippocampal and amygdalar function.
The first study from my lab led to a major discovery in the field of stress neurobiology (Vyas et al., J. Neurosci., 2002). Consistent with earlier reports, we found chronic stress to cause dendritic atrophy in hippocampal CA3 pyramidal neurons. Surprisingly, the same stress had the opposite effect on the basolateral amygdala (BLA) in the same brain – it triggered dendritic growth and spinogenesis in excitatory principal neurons of the BLA. Interestingly, only those forms of chronic stress that enhanced anxiety-like behavior also caused amygdalar spinogenesis and dendritic hypertrophy. In addition to providing the first evidence for structural plasticity induced in the adult amygdala by an aversive experience, our results raised the possibility that certain forms of chronic stress, by affecting specific neuronal elements in the amygdala, lead to behavioral manifestations of enhanced emotionality. Over the past 15 years, we have explored the functional implications of these findings using a range of electrophysiological, molecular, and behavioral assays. For instance, using single-cell recordings we found that Long-Term Potentiation (LTP) and NMDA receptor-mediated synaptic responses are also enhanced in the amygdala (Suvrathan et al., Phil. Trans. Royal Soc., 2014). Together, these synaptic changes enable amygdalar neurons to undergo further experience-dependent modifications, leading to stronger fear memories (Fig. 1). Our work showed how stress leaves its mark in the amygdala by generating new synapses with greater capacity for plasticity, thereby creating an ideal neuronal substrate for affective disorders. Importantly, these findings also highlighted for the first time the unique features of stress-induced plasticity in the amygdala that are strikingly different from the stress-induced deficits in the hippocampus and medial prefrontal cortex.
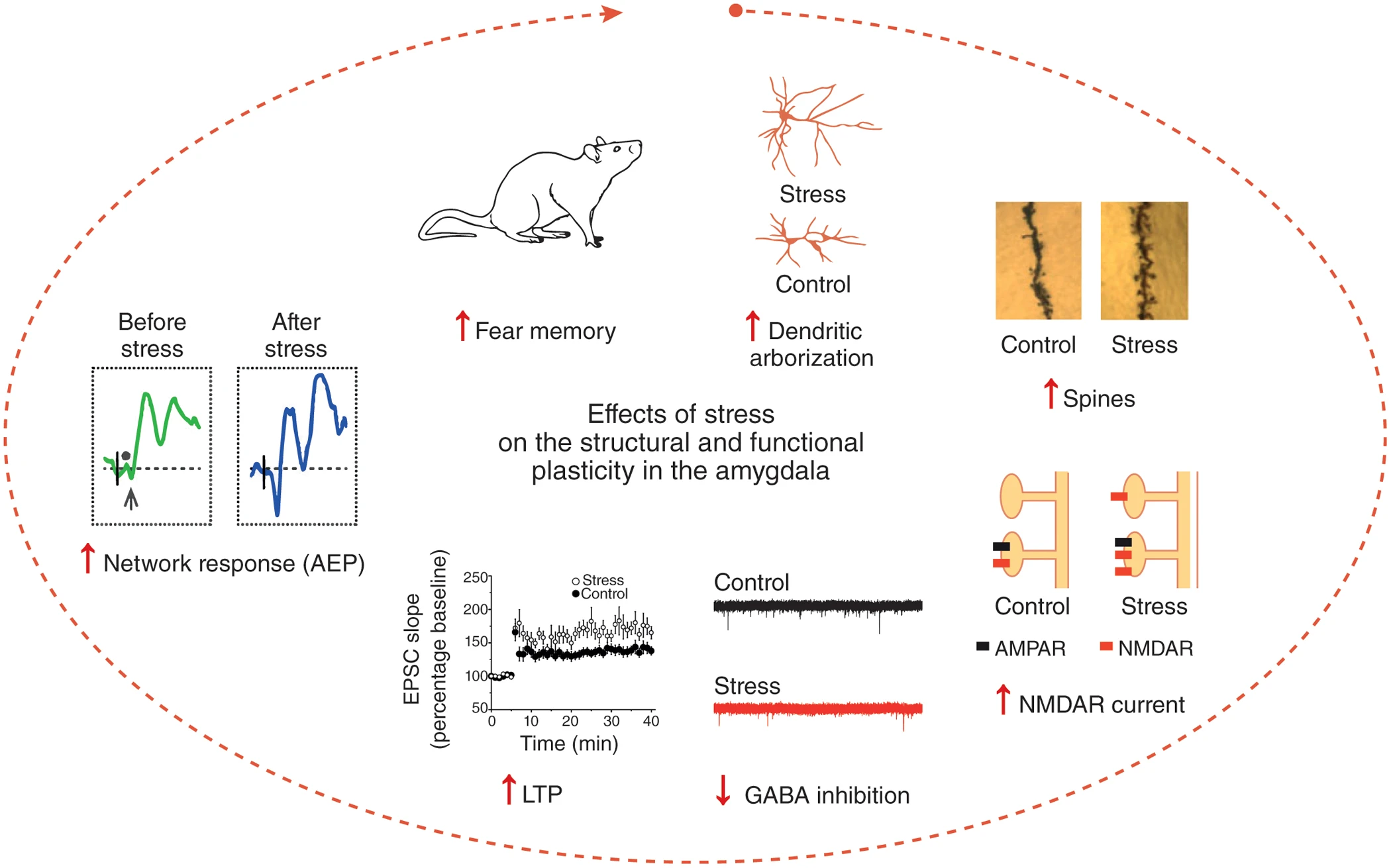
Fig 1: Stress enhances fear by forming new synapses with greater capacity for LTP in the lateral amygdala. Chronic stress strengthens the structural basis of synaptic connectivity causing dendritic growth and spinogenesis. These newly formed dendritic spines have larger NMDAR-mediated synaptic currents as a result of the formation of NMDAR-only or silent synapses. Stress also lowers synaptic inhibition. This creates conditions that facilitate the induction of greater LTP in the amygdala. This, in turn, gives rise to stronger auditory-evoked potentials (AEPs) in awake, behaving animals. Together, these cellular and network level changes give rise to stronger fear memories (from Chattarji et al., Nature Neuroscience, 2015)
Future directions: Our work in this area is now focusing on elucidating the molecular underpinnings of these brain-region specific differences in stress-induced plasticity.
1.2 Delayed effects of a single episode of stress – from animal models to therapeutic strategies
A majority of earlier studies using rodent models of repeated stress focused on the immediate, short-term effects of stress and may not elucidate key temporal features of certain types of stress disorders in humans. For example, post-traumatic stress disorder (PTSD) is triggered by a single overwhelmingly traumatic, often life-threatening, event. Strikingly, some components of the fear response in PTSD persist well beyond the original traumatic event, and may not even become evident till later in life. Hence, we initiated several studies aimed at capturing some of the defining temporal features of PTSD by building upon an earlier study wherein we reported (Fig. 2) that a single 2-hour episode of acute immobilization stress caused higher anxiety and formation of dendritic spines in the BLA not one, but ten days later (Mitra et al., PNAS, 2005; Roozendaal et al., Nature Rev. Neurosci., 2009).
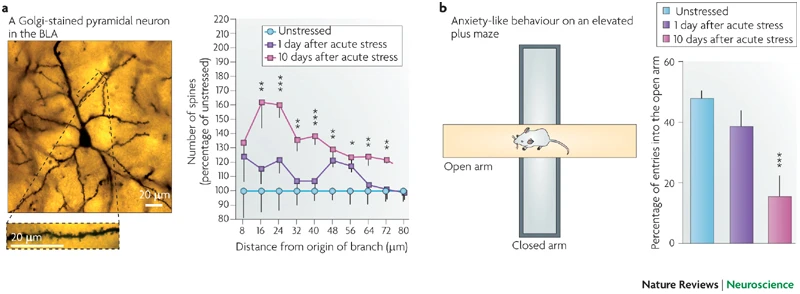
Fig. 2: A brief exposure to stress triggers a delayed increase in anxiety and spine density in the BLA (from Roozendaal, McEwen, and Chattarji. Nature Reviews Neuroscience, 2009)
One of the most counterintuitive features of PTSD comes from clinical reports showing that cortisol treatment reduces the cardinal symptoms of PTSD. We subjected our rodent model of PTSD to the challenge of replicating these clinical findings (Rao et al., Biol. Psychiatry, 2012). First, in a series of hormone depletion and replacement studies using adrenalectomized rats, we find that corticosterone indeed prevents stress-induced increase in anxiety. Second, an excess of corticosterone provided in the drinking water before acute stress completely blocked the delayed increase in anxiety. Further, in both of these experiments, corticosterone prevented stress-induced spinogenesis in the BLA, thereby confirming the correlation between stress-induced spinogenesis in the amygdala and high anxiety. Taken together, these results provide novel mechanistic insights into the protective, albeit puzzling, role for corticosterone in the modulation of stress-induced anxiety.
Future directions: Strikingly, ongoing work in our lab suggests that oral administration of corticosterone even a day after the same stress also prevents the delayed increase in anxiety and amygdalar spines ten days later. Therefore, the mechanisms and characteristics of post-stress interventions is a major focus of our current work.
[Collaborator: Bruce McEwen, Rockefeller University, USA]
1.3 Preventing the delayed impact of acute stress on the amygdala
Using whole-cell recordings from excitatory neurons in lateral amygdalar (LA) slices, we find that the same form of acute stress (described above) causes a significant increase in the frequency of miniature excitatory postsynaptic currents (mEPSCs) ten days later. Enhanced pre-synaptic release of glutamate at thalamic inputs to lateral amygdala neurons contributes to this increase. Further, targeted infusion of an NMDA-receptor antagonist into the basolateral amygdala during acute stress prevents both the increase in mEPSC frequency and spine-density ten days later (Yasmin et al., Physiol. Reports, 2017). Recently, we have also identified a role for the endocannabinoid (eCB) system, which is involved in modulation and gating of pre-synaptic neurotransmitter release. Oral administration of a pharmacological inhibitor of fatty acid amide hydrolase (FAAH), an enzyme involved in hydrolyzing eCB, before acute stress prevents the delayed strengthening of structural and physiological connectivity in the amygdala. These findings are consistent with accumulating clinical evidence for an important role played by the eCB system in protecting against PTSD and suggest pharmacological modulation of the eCB system as a possible therapeutic target.
Future directions: We are now investigating if post-stress inhibition of FAAH activity is also effective in reversing the delayed effects of acute stress. We are also probing the potential links between the eCB system and cortisol treatment in preventing the delayed effects of stress on amygdalar function.
[Collaborators: Bruce McEwen, Rockefeller University, USA; Matt Hill, University of Calgary, Canada]
2. Effects of stress on the intact brain in awake, behaving animals: from cells to networks
In earlier studies, stress-induced plasticity in different brain regions was viewed as stand-alone effects manifested as properties intrinsic to individual structures. Further, function was inferred from analysis at the cellular and behavioral levels without any online readout of dynamic changes in neuronal activity in the intact animal. However, neuroanatomical data also points to extensive interconnections between the hippocampus, medial prefrontal cortex (mPFC) and amygdala. This raises the intriguing possibility that some of the structural and physiological changes triggered by stress in one brain area may, at least in part, influence changes in other areas. Therefore, we are using in vivo recordings in freely behaving animals to examine the potential interdependence and interactions between brain areas differentially affected by stress.
2.1 Functional connectivity from the amygdala to the hippocampus grows stronger after stress
Our earlier work showed that the cellular and molecular effects of stress on the amygdala are strikingly different compared with those in the hippocampus. However, little is known about how stress affects dynamic changes and interactions in neuronal activity between the two areas. Hence, we have returned to the same basic question that inspired the first studies in my lab – but, with the more powerful tools of in vivo electrophysiological recordings in awake, behaving rats, wherein we simultaneously monitored electrical activity (Fig. 3) of neuronal populations located in hippocampal areas CA1 and CA3 and the LA during and after chronic immobilization stress (Ghosh et al., J. Neurosci., 2013). The amplitude of auditory-evoked potentials (AEPs) in the hippocampus increased transiently only after a single 2 h stress but not when it was repeated for 10 d. In contrast, both acute and chronic stress caused a persistent increase in AEPs in the LA. Chronic stress also elicited a sustained increase in the LA but a decrease in the hippocampus in the evoked power of gamma and beta frequencies. Moreover, beta and gamma synchrony was reduced between areas CA1 and CA3 but enhanced between the LA and hippocampus after chronic stress. Granger causality spectra revealed a strong directional influence from the LA to areaCA1that persisted throughout and even 10 d after chronic stress. However, directional coupling from hippocampal area CA3 to CA1 became weaker at the end of chronic stress. Thus, our findings suggest that the growing dominance of amygdalar activity over the hippocampus during and even after chronic stress may contribute to the enhanced emotional symptoms, alongside impaired cognitive function, seen in stress-related psychiatric disorders.
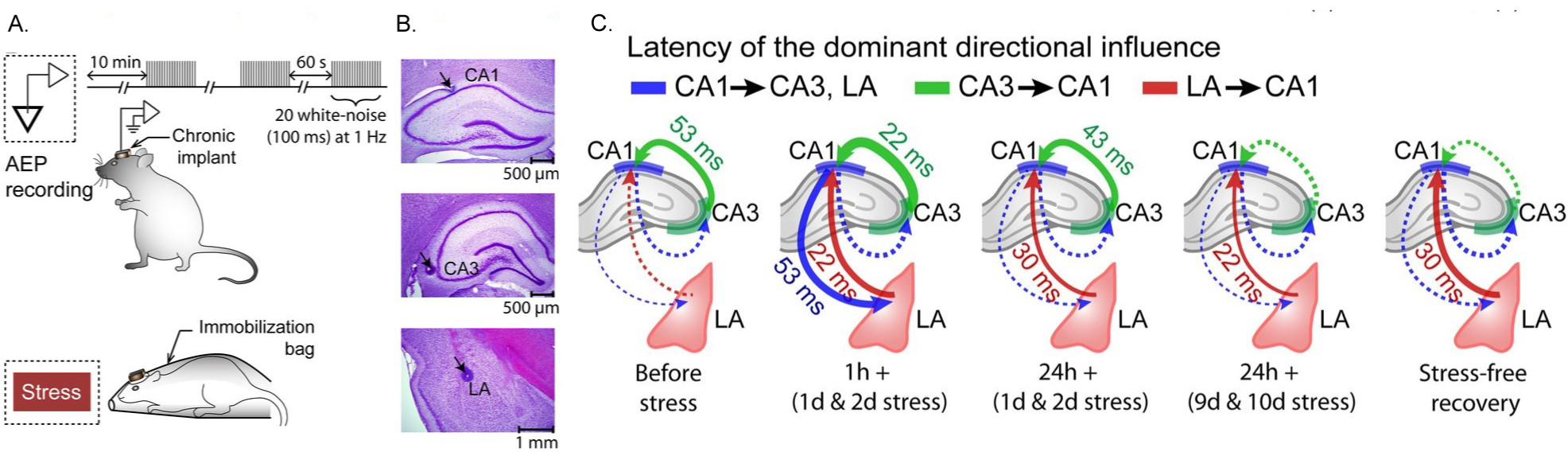
Fig 3: (A) Chronic in vivo recordings of AEPs simultaneously from hippocampal areas CA1 and CA3 and the (LA) in the same animal before, during, and after chronic stress. (B) Histological verification of recording sites in hippocampal areas CA1 and CA3 and the LA. (C) Granger causality graphs depicting the modulation of directional influence during different stages of stress between the LA and areas CA1 and CA3. The strength of Granger spectral causality values are coded by the thickness of lines. Solid and dotted lines indicate presence and absence of dominant directional influence, respectively (from Ghosh, Rao & Chattarji, J. Neurosci, 2013)
Future directions: We are extending these analyses to investigate how dynamic interactions between the amygdala and medial prefrontal cortex (mPFC) are influenced by chronic stress while the animal undergoes formation and extinction of fear memories.
2.2 The Dynamic Impact of Repeated Stress on the Hippocampal Spatial Map
Stress alters the function of many physiological processes throughout the body, including in the brain. A neural circuit particularly vulnerable to the effects of stress is the hippocampus, a key component of the episodic and spatial memory system in both humans and rodents. Earlier studies have provided snapshots of morphological, molecular, physiological and behavioral changes in the hippocampus following either acute or repeated stress. However, the cumulative impact of repeated stress on in vivo hippocampal physiology remains unexplored. Here we report (Tomar et al., Hippocampus, 2014) the stress-induced modulation of the spatially receptive fields of the hippocampal CA1 ‘place cells’ as mice explore familiar and novel tracks after 5 and 10 days of immobilization stress. We find that similar to what has been observed following acute stress, five days of repeated stress results in decreased excitability of CA1 pyramidal cells. Following ten days of chronic stress, however, this decreased hippocampal excitability is no longer evident, suggesting adaptation may have occurred. In addition to these changes in neuronal excitability, we find deficient context discrimination, wherein both short-term and chronic stress impair the ability of the hippocampus to unambiguously distinguish novel and familiar environments. These results suggest that a loss of network flexibility may underlie some of the behavioral deficits accompanying chronic stress.
We also noted that relatively little is known about the gradual changes in hippocampal structure, and its behavioral consequences, over the course of repeated stress. Our behavioral analyses during 10 days of chronic stress pointed to a delayed decline in spatial memory, the full impact of which is evident only after the end of stress. In contrast, concurrent volumetric measurements in the same animals revealed significant reduction in hippocampal volumes in stressed animals relative to their unstressed counterparts, as early as the third day of stress. Notably, animals that were behaviorally the worst affected at the end of chronic stress suffered the most pronounced early loss in hippocampal volume. Together, these findings (Rahman et al., Scientific Reports, 2016) support the view that not only is smaller hippocampal volume linked to stress-induced memory deficits, but it may also act as an early risk factor for the eventual development of cognitive impairments seen in stress-related psychiatric disorders.
[Collaborator: Thomas McHugh, RIKEN Brain Science Institute, Japan]
2.3 To be or not to be afraid: neuronal encoding of generalized fear in the amygdala
One of the most powerful model systems for elucidating the neural basis of learning is Pavlovian fear conditioning, in which subjects rapidly learn to associate a previously neutral tone (CS, conditioned stimulus) with a coincident aversive stimulus (US, unconditioned stimulus), e.g. a foot-shock. Re-exposure to the CS alone elicits a conditioned response — “freezing”, which provides a measure for the learned association. The early stages of fear memory formation involve strengthening of sensory afferents from the thalamus to the lateral amygdala (LA). As useful as this simple behavioral model has been in studying basic cellular mechanisms of associative learning, it does not capture some of the essential features of learning in the “real world”, where learned associations are rarely invariant over time and need to be generalized appropriately to novel settings. There are costs associated with both too little and too much generalization. Understanding these processes is also critical for gaining insights into psychiatric disorders such as post-traumatic stress disorder (PTSD), which may be viewed as an instance of overgeneralization.
Neuronal encoding of the transition from cue-specific to generalized fear is poorly understood. Hence, we used in vivo unit recordings in awake, behaving rats to examine the neuronal encoding of fear generalization (Ghosh & Chattarji, Nature Neurosci., 2015). We identified distinct neuronal populations in the lateral amygdala (LA) of rats that signaled generalized versus cue-specific associations and determined how their distributions switched during fear generalization. Notably, the same LA neurons that were cue-specific before the behavioral shift to generalized fear lost their specificity afterwards, thereby tilting the balance of activity toward a greater proportion of generalizing neurons. Neuronal activity in the LA, but not the auditory cortex, was necessary for fear generalization. Furthermore, targeted activation of cAMP/PKA signaling in the LA increased neuronal excitability of LA neurons and led to generalized fear. These results provide a cellular basis in the amygdala for the alteration of emotional states from normal to pathological fear.
Future Directions: We are building upon these findings to gain better insights into how these neuronal processes are affected in animal models of stress and autism spectrum disorders.
3. Autism Spectrum Disorders and the Amygdala
3.1 Characterization & reversal of synaptic defects in the amygdala in Fragile X Syndrome
While much of my earlier research focused on the neural basis of stress-induced modulation of fear and anxiety, the framework emerging from these studies have also led me to explore another class of disorders that too is characterized by severe cognitive and affective dysfunction – Fragile X Syndrome (FXS), the leading identified cause of autism. My initial work in this field, done in collaboration with Susumu Tonegawa (Hayashi et al., PNAS, 2007) and Mark Bear (Dolen et al., Neuron, 2007) at MIT, showed for the first time that it is possible to reverse or prevent many of the synaptic and behavioral phenotypes of FXS using genetic rescue strategies in mouse models. As valuable as these findings have been in validating or identifying molecular targets for therapeutic interventions against FXS, they had certain limitations. For example, our findings on genetic rescue were derived mainly from studies in the hippocampus and cortex, brain areas involved in cognitive function. However, FXS is also associated with strong emotional symptoms, which are likely to involve changes in the amygdala. Unfortunately, the synaptic basis of amygdalar dysfunction in FXS remained largely unexplored till the first analysis done in my lab (Suvrathan et al., PNAS, 2010; Suvrathan & Chattarji, Curr. Opinion Neurobiol., 2011). We identified synaptic defects in the basolateral amygdala that are in many respects distinct from those reported earlier in the hippocampus. Furthermore, despite the contrasting manifestations of abnormal synaptic transmission and plasticity in the amygdala versus hippocampus, our results suggested that synaptic defects in the amygdala of Fmr1-KO mice are still amenable to pharmacological interventions against mGluR5, albeit in a manner not envisioned in the original framework of the mGluR theory proposed by earlier research in the field.